Graduate student Leah Gulyas explores how one tiny coronavirus protein blockades host cells—and how a few simple changes can flip the script.
Mission control! Mission control! Do you read me? While these lines might first bring to mind a movie scene of astronauts frantically trying to contact their home base following a disastrous event in space, they might equally be applied to something a little closer to home—your own cells.
You can think of your cells like a little hive of activity, with proteins and other biomolecules going about their duties, working to keep the unit strong and healthy. And the main overseer—the mission control, if you will—is the cell’s nucleus. The nucleus sends a constant stream of messages in the form of RNA out into the cell, giving instructions and coordinating activity to keep everything running smoothly.
But what happens when something goes wrong? What happens when invaders come knocking? Say, perhaps a virus? Now the cell has a threat from the outside to contend with, and mission control must respond…if it can.
Communication breakdown
As a third-year PhD student in Britt Glaunsinger’s lab at UC Berkeley, I spend a lot of my time thinking about the complex interactions between a host and virus. During viral infection, your cells become a battleground, with the insidious virus subverting the host cell defenses in order to copy itself and make a new army of viruses. When that battalion is big enough, the viruses explode out of the cell and go on to further invasion.
Of course, your cells aren’t just bystanders in this process. The host cell also has tactics to prevent the enemy incursion. Our mission control (the nucleus) springs into action, transcribing commands (in the form of messenger RNA, or mRNA for short) that are sent to the field (the cytoplasm) to be translated into cellular activities to fight the virus. Literally, these messages are decoded by the protein-synthesizing machinery in the cell, the ribosome, and used for instruction to churn out new equipment (proteins) that restrict the viral infection. You might breathe a sigh of relief at this coordinated response—surely that will be enough to stop the invaders, right?
Well, unfortunately the enemy is cunning, and it has evolved over time to know just how to take advantage of its adversary. The enemy is going to do something very clever—it’s going to sever communication from mission control.
Scientists call this process “host shutoff.” It can happen in a number of a ways, but it’s a common feature of many viruses and has some hefty consequences for the host cell. Without instructions from the nucleus being carried out, the cell has difficulty preventing the virus from a takeover. This feature of infection and its downstream impacts have been a major area of research in the Glaunsinger Lab for many years now, but its effects recently became much more tangible as the coronavirus, SARS-CoV-2, hit the scene. As it happens, CoV-2 has its own strategies for shutting off the host, using a protein called nsp1, and this mechanistic aspect was dissected in a recent paper published in Cell Reports. This work was a collaborative effort between the Glaunsinger, Jamie Cate, and Nicholas Ingolia labs of UC Berkeley, originating during the statewide lockdown, and continuing to inspire interesting research questions today.
I spoke to my lab mates and advisor to get an inside view of what work on this project entailed.
Intercept and destroy
Host shutoff by coronaviruses wasn’t unknown at the start of the pandemic; in fact, our lab had previously worked with one of the main CoV proteins responsible (you might call it a soldier), Non-Structural Protein 1 (nsp1).
What is nsp1 and what did scientists already know about it? Postdoctoral fellow Aaron Mendez puts it this way: “It’s one of the first proteins synthesized by SARS-CoV, and it has this effect of blocking or interfering with host translation. It’s a very interesting protein in the sense that it renders this huge mega-Dalton protein complex [the ribosome] relatively inactive. It’s kind of like the equivalent of throwing a penny at a car and all of a sudden the car stops.”
Glaunsinger, my advisor, elaborates with another analogy: “nsp1 takes part of its protein domain and inserts that domain into the messenger RNA entry channel of the ribosome. This is what allows RNAs to be translated. Nsp1 acts like a cork in a wine bottle, stoppering the entry channel up so the mRNA can’t access the ribosome.”
Essentially, the miniscule nsp1 clogs the massive cellular machinery to make proteins and prevents our cells from generating the tools it needs to defeat its viral foe. Nsp1 also goes a step further to halt host messages from getting through; it helps to cut up the messages and destroy them, so they can’t simply get translated into protein by other ribosomes.
However, we didn’t have a complete picture of how nsp1 was performing its various roles. Nsp1 is a protein with three different parts, called domains, and two of them weren’t well-characterized. The researchers naturally wondered how they might be involved, if at all.
“The main focus was trying to dissect the roles of the N-terminal and central domain to characterize if they have any functional role in the ability of nsp1 to perform host shutoff,” says fellow graduate student Michael Ly. The group did this by swapping out what they suspected were important areas (residues) of these domains to determine if the modifications impacted host shutoff. “My main role was characterizing the effects of these mutations on nsp1,” continues Ly. “What we found was these two domains contribute to the main goal of nsp1 in blocking translation.”
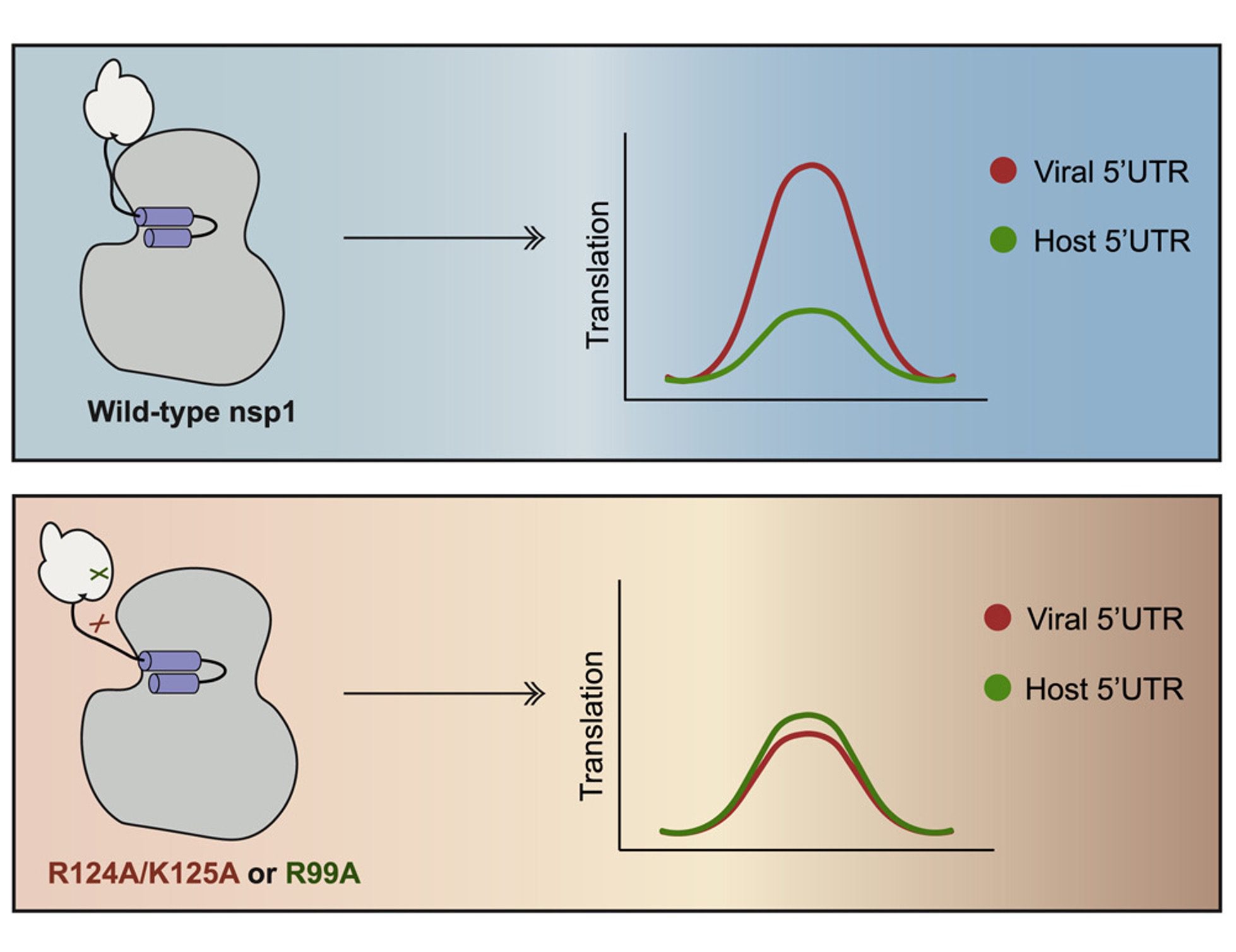
We now know that all three domains of nsp1 are important for preventing the nucleus from getting its messages out during infection. But there’s another twist—those ribosomes that nsp1 is so busy jamming up? They aren’t just something the host needs. To generate a massive viral army, CoV-2 relies on those same ribosomes to pump out the equipment it requires—but how can it do that with its own nsp1 getting in the way?
Enemy propaganda
Unsurprisingly, the enemy virus has developed ways to circumvent the problem of nsp1 repression. It turns out the same regions of the nsp1 protein that the researchers found to play a major role in host shutoff had another, unsuspected function during infection.
“They are important for allowing successful production of protein from viral transcripts” explains Ella Hartenian, a former graduate student in the lab. “This region [of nsp1] is critical for how it is differentiating the fate of host and viral transcripts.”
You see, CoV-2 messenger RNAs have a special feature in the beginning of the transcript called a “leader sequence.” You can think of it like a little signal that identifies the message as coming from the virus. This special sequence seems to tell nsp1 that the corresponding message is one the virus wants translated into protein. nsp1 can then act a bit like a bouncer, blocking host messages from getting in and freeing up all the ribosomal real-estate specifically for the viral messages it recognizes. The outcome? nsp1 steals the host’s resources to propagate its own enemy messages.
Intriguingly, my lab mates revealed that nsp1’s ability to perform this host versus viral message sorting is a precarious balancing act. With changes in just a few key parts of the protein, nsp1 seems to become confused and the virus’ edge crumbles. Mendez describes how, without those parts of the nsp1, “the host has the advantage, it is able to modestly access the ribosome, but viral sequences are down. Now the tables have turned.”
The N-terminal and central domains of nsp1, then, are important for repressing host translation and allowing viral messages to get through. But like a switch getting flipped, losing activity in those pieces suddenly allows host but not viral transcripts to get translated. “It’s an Achilles’ heel for the virus,” Glaunsinger asserts.
Know your enemy
You might wonder if, by understanding how the virus works, we can exploit that Achilles’ heel to cripple it. For Glaunsinger, “One of the driving reasons to study this particular protein was that we knew from other people’s work with other coronaviruses that nsp1 is a really important virulence factor. It’s critical for coronaviruses to cause disease and evade innate immune detection.”
With the discovery of this nsp1 switch, she suggests that “instead of nsp1 working against the host, you can engineer it to work against the virus. Maybe you could design an antiviral molecule that will bind in a way that mimics those types of mutants, where now you’re preventing the host gene expression shutdown and preferentially downregulating viral gene expression.”
In addition to potential medical relevance, we can also learn a lot about ourselves by getting to know the enemy. A favorite expression of Glaunsinger’s is “viruses are not inventors, they are thieves.” If that’s the case, where did the virus pick up its nsp1 stratagem? How did the enemy operatives learn that a leader sequence can evade ribosomal repression? Are there similar types of messages used by host mission control that the virus imitated? In trying to answer these questions, we may further penetrate the inner machinations of our very own cells.
The allied forces
While it was incredible to hear about the science from this talented group of people, I also wanted to know what it was like work on nsp1 amidst the CoV-2 pandemic. When I asked Glaunsinger how the project got started, she replied, “Everybody was looking for ‘What’s the type of research I can do that will feel impactful during the pandemic?’ So, you’re sort of looking in your wheelhouse of skills that you have laboratory-wise, and this was a clear avenue we could take.”
Hartenian spent her graduate career in “the wheelhouse” of host shutoff and recalls that she was already somewhat familiar with nsp1. “But it wasn’t like I really knew it in all that much detail; it involved a lot of getting up to speed and reading literature.” However, she felt well-positioned for deeper study of nsp1. “The experience of working with host shutoff allowed you to synthesize and read the literature in a more efficient way and with an eye to the questions you would ask in a system you’re more familiar with.”
As research progressed during the pandemic, it was often challenging for the group to juggle nsp1 studies while maintaining their own research, according to Mendez, and this inevitably caused stress at times. But he points out that “we were all excited to be doing something that had a directionality towards CoV-2 and helping in some way.”
Ly agrees that it was nice to contribute to the coronavirus research in hopes that it could lead to better antivirals or preparation for another pandemic. However, these kinds of consequences are often distant, so he also participated in other activities. “One thing I did during the pandemic that felt more closely tied to the relevance of the pandemic was making hand sanitizers.” Like many others who unexpectedly became experts at making masks and home-brewing cleaning supplies, these direct impacts on surrounding communities helped him feel more engaged.
Altogether, the story of nefarious nsp1 and the surprising mutations that can bring it down is another intriguing piece of the host-viral conflict. As scientists piece more and more of CoV-2’s battle plan together, it will hopefully ensure that we—the hosts—can continue to strike back.