Sample Preparation Guidelines
Guidelines, protocols, and examples are given in the following references:
- “Overview of Affinity Purification.” (Pierce/Thermo)
- Dunham, W. H.; Mullin, M.; Gingras, A.-C. “Affinity purification coupled to mass spectrometry: Basic principles and strategies.” Proteomics 2012, 12, 1576-1590.
- Pflieger, D.; Bigeard, J.; Hirt, H. “Isolation and characterization of plant protein complexes by mass spectrometry.” Proteomics 2011, 11, 1824-1833.
- Paul, F. E.; Hosp, F.; Selbach, M. “Analyzing protein-protein interactions by quantitative mass spectrometry.” Methods 2011, 54, 387-395.
- Malovannaya, A.; Li, Y.; Bulynko, Y.; Jung, S. Y.; Wang, Y.; Lanz, R. B.; O’Malley, B. W.; Qin, J. “Streamlined analysis schema for high-throughput identification of endogenous protein complexes.” Proc. Natl. Acad. Sci. USA 2010, 107, 2431-2436.
Examples are given in the following references:
- Offenbacher, A. R.; Hu, S.; Poss, E. M.; Carr, C. A. M.; Scouras, A. D.; Prigozhin, D.; Iavarone, A. T.; Palla, A.; Alber, T.; Fraser, J. S.; Klinman, J. P. “Hydrogen-deuterium exchange of lipoxygenase uncovers a relationship between distal, solvent exposed protein motions and the thermal activation barrier for catalytic proton-coupled electron tunneling.” ACS Cent. Sci. 2017, 3, 570-579.
- Underbakke, E. S.; Iavarone, A. T.; Marletta, M. A. “Higher-order interactions bridge the nitric oxide receptor and catalytic domains of soluble guanylate cyclase.” Proc. Natl. Acad. Sci. USA 2013, 110, 6777-6782.
- Tsutsui, Y.; Wintrode, P. L. “Hydrogen/deuterium exchange-mass spectrometry: A powerful tool for probing protein structure, dynamics and interactions.” Current Medicinal Chemistry 2007, 14, 2344-2358.
Guidelines, protocols, and examples are given in the following references:
- Thomas, S. L.; Thacker, J. B.; Schug, K. A.; Maráková, K. “Sample preparation and fractionation techniques for intact proteins for mass spectrometric analysis.” J. Sep. Sci. 2021, 44, 211-246.
- Donnelly, D. P. et al. “Best practices and benchmarks for intact protein analysis for top-down mass spectrometry.” Nat. Methods 2019, 16, 587-594.
- Wang, H.; Hanash, S. “Intact-protein based sample preparation strategies for proteome analysis in combination with mass spectrometry.” Mass Spectrom. Rev. 2005, 24, 413-426.
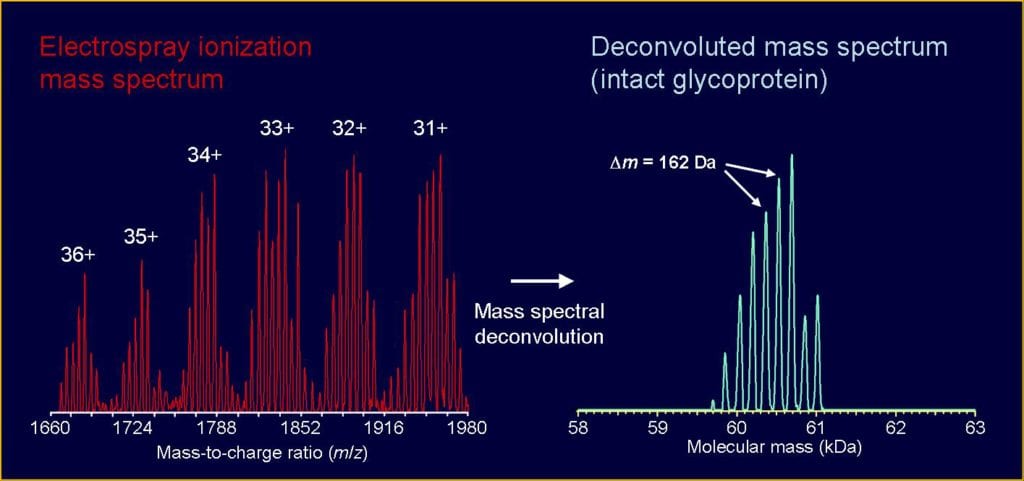
Guidelines, protocols, and examples are given in the following references:
- Saini, R. K.; Prasad, P.; Shang, X.; Keum, Y.-S. “Advances in lipid extraction methods—a review.” Int. J. Mol. Sci. 2021, 22, 13643.
- Aldana, J.; Romero-Otero, A.; Cala, M.P. “Exploring the lipidome: Current lipid extraction techniques for mass spectrometry analysis.” Metabolites 2020, 10, 231.
- Cajka, T.; Fiehn, O. “Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry.” TrAC, Trends Anal. Chem. 2014, 61, 192-206.
Guidelines, protocols, and examples are given in the following references:
- Raterink, R.-J.; Lindenburg, P. W.; Vreeken, R. J.; Ramautar, R.; Hankemeier, T. “Recent developments in sample-pretreatment techniques for mass spectrometry-based metabolomics.” TrAC, Trends Anal. Chem. 2014, 61, 157-167.
- Dettmer, K., Aronov, P. A.; Hammock, B. D. “Mass spectrometry-based metabolomics.” Mass Spectrom. Rev. 2007, 26, 51-78.
- Villas-Bôas, S. G.; Mas, S.; Åkesson, M.; Smedsgaard, J.; Nielsen, J. “Mass spectrometry in metabolome analysis.” Mass Spectrom. Rev. 2005, 24, 613-646.
Guidelines, protocols, and examples are given in the following references:
- Lorenzen, K.; Duijn, E. “Native mass spectrometry as a tool in structural biology.” Current Protocols in Protein Science 2010, 62, 17.12:17.12.1-17.12.17.
- Heck, A. J. R. “Native mass spectrometry: A bridge between interactomics and structural biology.” Nature Methods 2008, 5, 927-933.
- Mehmood, S.; Allison, T. M.; Robinson, C. V. “Mass spectrometry of protein complexes: From origins to applications.” Annu. Rev. Phys. Chem. 2015, 66, 453-474.
- Liu, T. Y.; Iavarone, A. T.; Doudna, J. A. “RNA and DNA targeting by a reconstituted Thermus thermophilus Type III-A CRISPR-Cas system.” PLoS ONE 2017, 12, e0170552.
For a detailed oligonucleotide sample preparation protocol, please see Shah, S.; Friedman, S. H. “An ESI-MS method for characterization of native and modified oligonucleotides used for RNA interference and other biological applications.” Nature Protocols 2008, 3, 351-356.
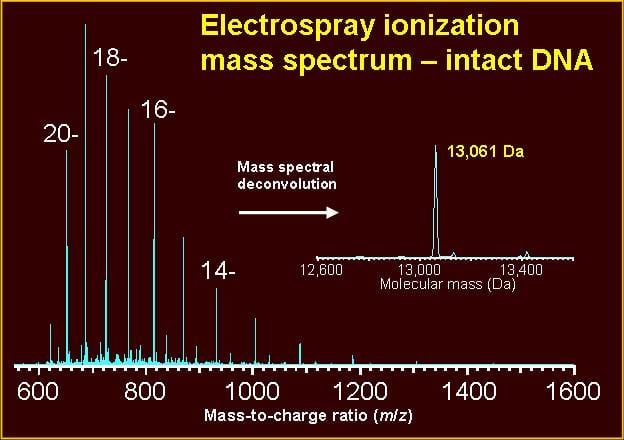
Guidelines, protocols, and examples are given in the following references:
- Strynar, M.; McCord, J.; Newton, S.; Washington, J.; Barzen-Hanson, K.; Trier, X.; Liu, Y.; Dimzon, I. K.; Bugsel, B.; Zwiener, C.; Munoz, G. “Practical application guide for the discovery of novel PFAS in
environmental samples using high resolution mass spectrometry.” J. Expo. Sci. Environ. Epidemiol. 2023, 33, 575-588. - Jia, S.; Marques Dos Santos, M.; Li, C.; Snyder, S. A. “Recent advances in mass spectrometry analytical techniques for per- and polyfluoroalkyl substances (PFAS).” Anal. Bioanal. Chem. 2022, 414, 2795-2807.
- Liu, Y.; D’Agostino, L. A.; Qu, G.; Jiang, G.; Martin, J. W. “High-resolution mass spectrometry (HRMS) methods for nontarget discovery and characterization of poly- and per-fluoroalkyl substances (PFASs) in environmental and human samples.” TrAC, Trends Anal. Chem. 2019, 121, 115420.
Guidelines, protocols, and examples are given in the following references:
- Wesdemiotis, C.; Williams-Pavlantos, K. N.; Keating, A. R.; McGee, A. S.; Bochenek, C. “Mass spectrometry of polymers: A tutorial review.” Mass Spectrom Rev. 2024, 43, 427-476.
- De Bruycker, K.; Welle, A.; Hirth, S.; Blanksby, S. J.; Barner-Kowollik, C. “Mass spectrometry as a tool to advance polymer science.” Nat. Rev. Chem. 2020, 4, 257-268.
- Nielen, M. W. F. “Maldi time-of-flight mass spectrometry of synthetic polymers.” Mass Spectrom. Rev. 1999, 18, 309-344.
Guidelines, protocols, and examples are given in the following references:
- Li, X.; Franz, T.; Atanassov, I.; Colby, T. “Step-by-step sample preparation of proteins for mass spectrometric analysis.” In: Posch, A. (ed.) Proteomic Profiling. Methods in Molecular Biology 2021, 2261, 13-23. Humana, New York, NY.
- Mansuri, M. S.; Bathla, S.; Lam, T. T.; Nairn, A. C.; Williams, K. R. “Optimal conditions for carrying out trypsin digestions on complex proteomes: From bulk samples to single cells.” J. Proteomics 2024, 297, 105109.
- Muriithi, B.; Ippoliti, S.; Finny, A.; Addepalli, B.; Lauber, M. “Clean and complete protein digestion with an autolysis resistant trypsin for peptide mapping.” J. Proteome Res. 2024, 23, 5221-5228.
- Jiang, Y. et al. “Comprehensive overview of bottom-up proteomics using mass spectrometry.” ACS Meas. Sci. Au 2024, 4, 338-417.
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J. V.; Mann, M. “In-gel digestion for mass spectrometric characterization of proteins and proteomes.” Nat. Protocols 2006, 1, 2856-2860.