
General Mass Spectrometry Protocols
Guidelines, protocols, and examples are given in the following references:
- “Overview of Affinity Purification.” (Pierce/Thermo)
- Dunham, W. H.; Mullin, M.; Gingras, A.-C. “Affinity purification coupled to mass spectrometry: Basic principles and strategies.” Proteomics 2012, 12, 1576-1590.
- Pflieger, D.; Bigeard, J.; Hirt, H. “Isolation and characterization of plant protein complexes by mass spectrometry.” Proteomics 2011, 11, 1824-1833.
- Paul, F. E.; Hosp, F.; Selbach, M. “Analyzing protein-protein interactions by quantitative mass spectrometry.” Methods 2011, 54, 387-395.
- Malovannaya, A.; Li, Y.; Bulynko, Y.; Jung, S. Y.; Wang, Y.; Lanz, R. B.; O’Malley, B. W.; Qin, J. “Streamlined analysis schema for high-throughput identification of endogenous protein complexes.” Proc. Natl. Acad. Sci. USA 2010, 107, 2431-2436.
Guidelines, protocols, and examples are given in the following references:
- Masson, G. R. et al. “Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments.” Nat. Methods 2019, 16, 595-602.
- James, E. I.; Murphree, T. A.; Vorauer, C.; Engen, J. R.; Guttman, M. “Advances in hydrogen/deuterium exchange mass spectrometry and the pursuit of challenging biological systems.” Chem. Rev. 2022, 122, 7562-7623.
- Offenbacher, A. R.; Hu, S.; Poss, E. M.; Carr, C. A. M.; Scouras, A. D.; Prigozhin, D.; Iavarone, A. T.; Palla, A.; Alber, T.; Fraser, J. S.; Klinman, J. P. “Hydrogen-deuterium exchange of lipoxygenase uncovers a relationship between distal, solvent exposed protein motions and the thermal activation barrier for catalytic proton-coupled electron tunneling.” ACS Cent. Sci. 2017, 3, 570-579.
Guidelines, protocols, and examples are given in the following references:
- Thomas, S. L.; Thacker, J. B.; Schug, K. A.; Maráková, K. “Sample preparation and fractionation techniques for intact proteins for mass spectrometric analysis.” J. Sep. Sci. 2021, 44, 211-246.
- Donnelly, D. P. et al. “Best practices and benchmarks for intact protein analysis for top-down mass spectrometry.” Nat. Methods 2019, 16, 587-594.
- Wang, H.; Hanash, S. “Intact-protein based sample preparation strategies for proteome analysis in combination with mass spectrometry.” Mass Spectrom. Rev. 2005, 24, 413-426.
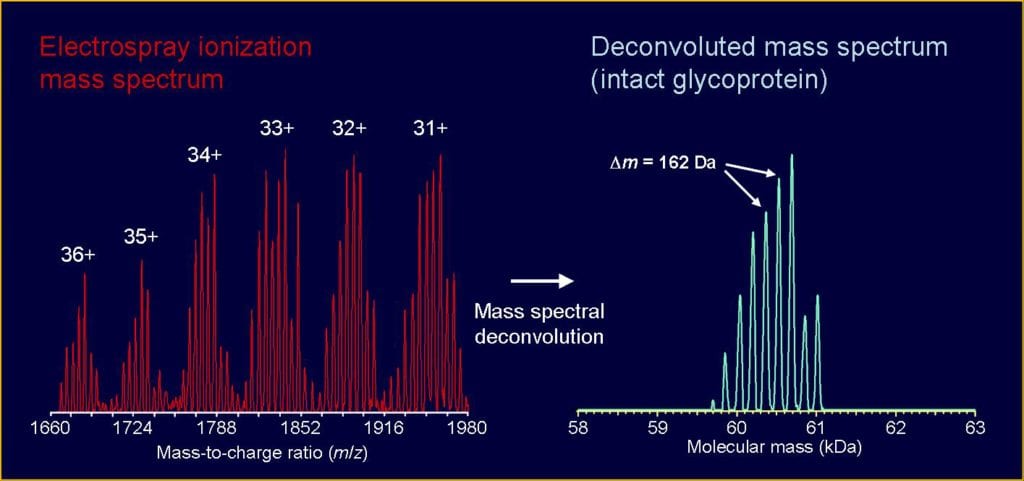
Guidelines, protocols, and examples are given in the following references:
- Saini, R. K.; Prasad, P.; Shang, X.; Keum, Y.-S. “Advances in lipid extraction methods—a review.” Int. J. Mol. Sci. 2021, 22, 13643.
- Aldana, J.; Romero-Otero, A.; Cala, M.P. “Exploring the lipidome: Current lipid extraction techniques for mass spectrometry analysis.” Metabolites 2020, 10, 231.
- Cajka, T.; Fiehn, O. “Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry.” TrAC, Trends Anal. Chem. 2014, 61, 192-206.
Guidelines, protocols, and examples are given in the following references:
- Raterink, R.-J.; Lindenburg, P. W.; Vreeken, R. J.; Ramautar, R.; Hankemeier, T. “Recent developments in sample-pretreatment techniques for mass spectrometry-based metabolomics.” TrAC, Trends Anal. Chem. 2014, 61, 157-167.
- Dettmer, K., Aronov, P. A.; Hammock, B. D. “Mass spectrometry-based metabolomics.” Mass Spectrom. Rev. 2007, 26, 51-78.
- Villas-Bôas, S. G.; Mas, S.; Åkesson, M.; Smedsgaard, J.; Nielsen, J. “Mass spectrometry in metabolome analysis.” Mass Spectrom. Rev. 2005, 24, 613-646.
Guidelines, protocols, and examples are given in the following references:
- Ayon, N. J. ”Preanalytical strategies for native mass spectrometry analysis of protein modifications, complexes, and higher-order structures.” AppliedChem 2025, 5, 35.
- Barth, M., Schmidt, C. “Native mass spectrometry—A valuable tool in structural biology.” J. Mass Spectrom. 2020, 55, e4578.
- Lorenzen, K.; Duijn, E. “Native mass spectrometry as a tool in structural biology.” Current Protocols in Protein Science 2010, 62, 17.12:17.12.1-17.12.17.
- Heck, A. J. R. “Native mass spectrometry: A bridge between interactomics and structural biology.” Nature Methods 2008, 5, 927-933.
Guidelines, protocols, and examples are given in the following references:
- Nuckowski, Ł.; Kaczmarkiewicz, A.; Studzińska, S. “Review on sample preparation methods for oligonucleotides analysis by liquid chromatography.” Journal of Chromatogr. B 2018, 1090, 90-100.
- Shah, S.; Friedman, S. H. “An ESI-MS method for characterization of native and modified oligonucleotides used for RNA interference and other biological applications.” Nat. Protocols 2008, 3, 351-356.
- Chou, C.-W.; Limbach, P. A. (2000), “Analysis of oligonucleotides by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry.” Current Protocols in Nucleic Acid Chemistry 2000, 00, 10.1.1-10.1.25.
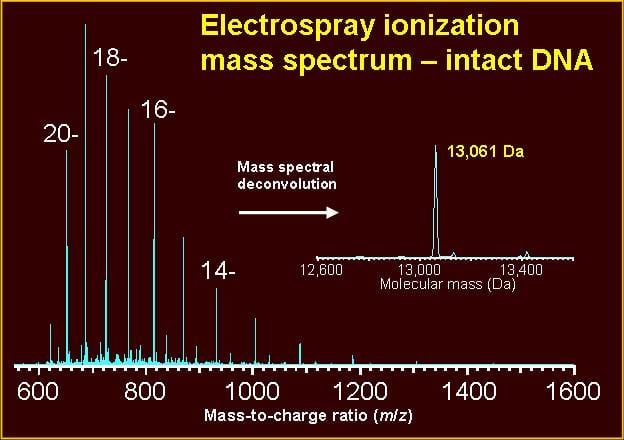
Guidelines, protocols, and examples are given in the following references:
- Strynar, M.; McCord, J.; Newton, S.; Washington, J.; Barzen-Hanson, K.; Trier, X.; Liu, Y.; Dimzon, I. K.; Bugsel, B.; Zwiener, C.; Munoz, G. “Practical application guide for the discovery of novel PFAS in environmental samples using high resolution mass spectrometry.” J. Expo. Sci. Environ. Epidemiol. 2023, 33, 575-588.
- Jia, S.; Marques Dos Santos, M.; Li, C.; Snyder, S. A. “Recent advances in mass spectrometry analytical techniques for per- and polyfluoroalkyl substances (PFAS).” Anal. Bioanal. Chem. 2022, 414, 2795-2807.
- Liu, Y.; D’Agostino, L. A.; Qu, G.; Jiang, G.; Martin, J. W. “High-resolution mass spectrometry (HRMS) methods for nontarget discovery and characterization of poly- and per-fluoroalkyl substances (PFASs) in environmental and human samples.” TrAC, Trends Anal. Chem. 2019, 121, 115420.
Guidelines, protocols, and examples are given in the following references:
- Wesdemiotis, C.; Williams-Pavlantos, K. N.; Keating, A. R.; McGee, A. S.; Bochenek, C. “Mass spectrometry of polymers: A tutorial review.” Mass Spectrom Rev. 2024, 43, 427-476.
- De Bruycker, K.; Welle, A.; Hirth, S.; Blanksby, S. J.; Barner-Kowollik, C. “Mass spectrometry as a tool to advance polymer science.” Nat. Rev. Chem. 2020, 4, 257-268.
- Nielen, M. W. F. “Maldi time-of-flight mass spectrometry of synthetic polymers.” Mass Spectrom. Rev. 1999, 18, 309-344.
Guidelines, protocols, and examples are given in the following references:
- Li, X.; Franz, T.; Atanassov, I.; Colby, T. “Step-by-step sample preparation of proteins for mass spectrometric analysis.” In: Posch, A. (ed.) Proteomic Profiling. Methods in Molecular Biology 2021, 2261, 13-23. Humana, New York, NY.
- Mansuri, M. S.; Bathla, S.; Lam, T. T.; Nairn, A. C.; Williams, K. R. “Optimal conditions for carrying out trypsin digestions on complex proteomes: From bulk samples to single cells.” J. Proteomics 2024, 297, 105109.
- Muriithi, B.; Ippoliti, S.; Finny, A.; Addepalli, B.; Lauber, M. “Clean and complete protein digestion with an autolysis resistant trypsin for peptide mapping.” J. Proteome Res. 2024, 23, 5221-5228.
- Jiang, Y. et al. “Comprehensive overview of bottom-up proteomics using mass spectrometry.” ACS Meas. Sci. Au 2024, 4, 338-417.
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J. V.; Mann, M. “In-gel digestion for mass spectrometric characterization of proteins and proteomes.” Nat. Protocols 2006, 1, 2856-2860.
Proteomics Protocols
LC-MS/MS data can be processed to detect any modifications that may be present in the peptides detected. The modification must add a calculable molecular mass. Modification analysis can be performed using either one-dimensional or multi-dimensional “MudPIT” chromatography depending on the sample complexity. We can often find modifications on a target protein even when it is part of a complex mixture. Often modifications of interest can be found simply by searching LC-MS data collected under standard conditions, but we can do better if the data collection is planned specifically with modification analysis in mind. For some types of modification including phosphorylation, we can collect additional data by CID, HCD or ETD fragmentation that confirms the presence of the modification with higher confidence. Ask us for details.
In some cases, chances of pinpointing the site of a modification increase if overlapping peptides are analyzed or if an enzyme other than trypsin is used to generate peptides. To produce sets of overlapping peptides, we recommend digestion with two or three proteases that vary in their specificity. If there is a suspected site of modification, the proteolytic digest can be planned to produce peptides of optimal mass containing that site. You can use peptidecutter to choose an appropriate protease.
To perform this type of analysis, please refer to the following forms and protocols:
- Lys-C or trypsin in-solution protein digest protocol
- TCA precipitation of proteins
- Sample desalting for mass spectrometry
- Sample submission form
We urge you to contact facility staff to discuss your project prior to sample preparation.
Nanoscale LC-MS/MS using CID, HCD or ETD fragmentation allows for the identification of individual peptides from a complex mixture. This is a powerful proteomics technique through which the component proteins in a sample can be identified.
Typically, a sample is prepared by tryptic digestion and desalting of a protein mixture obtained in the course of the user’s research. Samples have varied in complexity from purified protein complexes to whole cell extracts.
We load a desalted, proteolysed mixture on a nanoscale HPLC column. These columns employ either reverse phase (one-dimensional) or a combination of ion exchange and reverse phase (two-dimensional or MudPIT) separation chemistries. The eluent from the LC column is subjected directly to tandem mass spectrometry (MS/MS), and the mass and fragmentation spectrum of major ions is recorded.
A computer program suite is then used to identify the peptides from the spectra that have been collected. It queries a sequence database for the appropriate organism, calculating theoretical fragmentation spectra for all possible peptides and comparing them to the data collected.
The final output for the user is a file listing each gene for which peptides were found in the data. Each peptide is listed along with statistics showing the quality of the data.
One-dimensional separations are appropriate for samples where the expected complexity is 1 to 10 proteins. Between 10 ng and 500 ng are needed for the analysis.
Two-dimensional “MudPIT” separations are appropriate for more complex mixtures. Between 100 ng and 10 µg are needed for analysis.
Users generally prepare a proteolysed, desalted sample for one-dimensional or two-dimensional analysis.
To perform this type of analysis, please refer to the following forms and protocols:
- Lys-C or trypsin in-solution protein digest protocol
- Sample desalting for mass spectrometry
Protocol revised June 2011
We recommend Gelcode Blue® Coomassie stain (Pierce) for detecting bands. This technique works for any band that can be seen by this stain. 0.1 to 0.2 micrograms of protein is ideal. Use a new scalpel or razor blade to cut out each band. Mince each band into <1-mm2 pieces and transfer to a clean microcentrifuge tube.
Caution: Many silver stains, including those claiming mass spec. compatibility, give poor results. Colloidal Coomassie stains such as that recommended above work well. If you feel compelled to stain with silver, then the band should be completely de-stained (no visible color) before starting this protocol.
Note: To avoid contaminants use only Milli-Q water (or better) and wear gloves throughout preparation. All reagents should be HPLC grade. Prepare all solutions fresh.
- Wash the gel pieces for 20 min. in 500 μL of 100 mM NH4HCO3. Discard the wash.
- Add 150 μL of 100 mM NH4HCO3 and 10 μL of 45 mM DTT. Incubate at 50 °C for 15 min.
- Cool to room temperature and add 10 μL of 100 mM iodoacetamide and incubate for 15 min. in the dark at room temp.
- Discard the solvent and wash the gel slice in 500 μL of a 50:50 mix of acetonitrile and 100 mM NH4HCO3 with shaking for 20 min. Discard the solvent.
- Add 50 μL of acetonitrile to shrink the gel pieces. After 10-15 min., remove the solvent and dry the gel fragments in a speed vac.
- Re-swell the gel pieces with 10 μL of 25 mM NH4HCO3 containing Promega modified trypsin (sequencing grade) at a concentration such that a substrate-to-enzyme ratio of 10:1 has been achieved. If the amount of protein is not known, then add 10-20 μL of 25 mM NH4HCO3 containing 0.1-0.2 μg of trypsin. After 10-15 min., add 10-20 μL of additional buffer, enough to cover the gel pieces. Incubate overnight (8 hours or more) at 37 °C.
- Remove the supernatant and place in new microcentrifuge tubes. Extract remaining peptides from the gel pieces twice with 50 μL of 60% acetonitrile/0.1% formic acid for 20 min, then once with 25 μL acetonitrile. Add these extracts to appropriate tubes containing the supernatant of the sample. Speed-vac to dryness.
Sample submission form
We urge you to contact facility staff to discuss your project prior to sample preparation.
Equipment:
- C18 Spec tips, Agilent, cat #A57203
- p200 and p1000 pipette
Reagents:
- HPLC grade methanol
- 5% HPLC grade acetonitrile/5% formic acid
- 80% HPLC grade acetonitrile/5% formic acid
- Make solutions with Milli-Q or better water. Note: Do not pipette concentrated formic acid with plastic pipette tips; use glass.
You can use a p1000 pipette to wash and elute spec tips. For each wash, add the required volume into the top of the spec tip, then press a p1000 tip firmly into the spec tip and depress the plunger to drive the liquid through the device.
- Wet the spec tip by pushing through 200 µL of HPLC grade methanol.
- Wash the spec tip with 3 x 200 µL 5% acetonitrile/5% formic acid.
- If your sample does not already contain an acidic ion pairing agent, add formic acid to your sample to 5%. NOTE: Do not pipette concentrated formic acid with plastic pipette tips; use glass.
- Push the sample through the spec tip. You may pass the sample through twice, washing with 5% acetonitrile/5% formic acid between each pass, if you would like to maximize binding.
- Wash with 3 x 200 µL 5% acetonitrile/ 5% formic acid.
- Elute with 2 x 100 µL 80% acetonitrile/5% formic acid.
- Speed vac the sample to dryness.
Original protocol from the J. Yates Lab, Scripps Research Institute.
Solutions:
1 M TCEP
for 1 mL:
287 mg
1 mL Milli-Q water
Make a 1/10 dilution and store at -20 °C in aliquots.
500 mM iodoacetamide
for 0.5 mL:
46 mg
500 µL ddH2O; make fresh.
1 M CaCl2
for 100 mL:
14.7 g CaCl2•2H2O
ddH2O to 100 mL
Filter sterilize.
- Bring solution up to 8 M urea and 100 mM Tris-HCl pH 8.5 (use 10 M urea and 1 M Tris stocks; ideal final volume is about 80 µL).
- Add 100 mM TCEP (a reducing agent) to a final concentration of 5 mM. Incubate at room temp. for 20 min (2.5 µL if the total volume was 80 µL).
- Add 500 mM iodoacetamide (make fresh daily) to a final concentration of 10 mM. Incubate at room temp. for 15 min. in the dark (covered with foil).
- Use one of the following enzymes (trypsin is recommended for MudPIT):
Trypsin Digest:
- Dilute samples by a factor of four with 100 mM Tris-HCl pH 8.5 (final urea conc. = 2 M; if sample volume is a problem, dilute by only a factor of two to 4 M urea).
- Add 100 mM CaCl2 to a final conc. of 1 mM.
- Add in 1 µL trypsin (0.5 µg/µL). Promega sequencing grade trypsin is recommended.
- Incubate overnight at 37 °C in the dark.
Lys-C Digest:
- Add in 1 µL Lys-C (0.1 µg/µL), 1/100th total amount.
- Incubate for 4 hr. at 37 °C in the dark.
- Add formic acid to 5% final conc.
Protocol prepared by Lori Kohlstaedt, June 2006; revised April 2008.
Preparation of 100% trichloroacetic acid (TCA):
(Don’t try to weigh out TCA; it’s too hygroscopic.)
- Obtain a fresh bottle of crystalline TCA.
- Read the weight in the container from the label.
- Add distilled water to give a 100 g / 100 mL solution at final volume. Store the solution in an acid-compatible container.
- Add 100% (w/v) TCA to the sample to bring the TCA concentration to 20%.
- Incubate on ice for at least 1 hr. Dilute samples may be left overnight.
- Spin at maximum speed at 4 degrees Celsius in a microcentrifuge for 10 min.
- Wash the pellet 3X with a solution of ice-cold 0.01 M HCl / 90% acetone.
- Allow the pellet to air dry.
The pellet can then be resuspended directly in 100 mM Tris, pH 8.5, 8 M urea for enzymatic digestion for mass spectrometry.
- Plan cell growth or IP so that the target sample will be 100 µg or more of protein.
- Take digested peptides either from a large-scale IP or a whole-cell lysate and desalt using C18 column and speed vac dry overnight. (See peptide desalting and trypsin digestion protocols.)
- Re-suspend peptides in 200 mM HEPPS pH 8.
- Quantify peptides using Pierce Quantitative Colorimetric Peptide Assay (Pierce 23275). (Fisher accuSkan FC use Abs 490 in mass spec facility.)
- Normalize peptides to be compared to equal concentration and volume in 200 mM HEPPS pH 8 final volume 100 µL for one reagent vial. (Can use less with ratio of 8 µg of TMT reagent to 1 µg/peptide; range recommended: 25-100 µg.)
- Label using TMT labeling reagents (protocol TMT Mass Tagging Kits and Reagents) (used TMT duplex Label Reagent Set)
- Before use equilibrate the TMT label reagents at room temp.
- Re-suspend .8 mg vial with 41 µL anhydrous acetonitrile. Allow reagent to dissolve for 5 minutes with occasional vortexing. Centrifuge tube briefly.
- Add 41 µL of re-suspended TMT reagent to tube of peptides (100 µL).
- Mix gently and let react for 1 hour at room temp.
- Quench reaction with 8 µL 5% hydroxylamine for 15 min.
- Combine samples at equal amounts in a new tube and store at -80 °C.
- Desalt using C18 column and speed vac dry.
- Mass spec time!